
지난 2023년, 미국 FDA 감사에서 무균 의약품 제조업체에 대해 무균 시설 설계, 오염 관리, 장비 세척/유지보수, 환경 모니터링 및 포괄적인 공정 밸리데이션 등의 문제가 강조되었다.
2023년 감사 대상 시설에서 위반한 상위 CFR Section(표 1)은 Section 211.42, 211.113 및 211.67이다. Section 211.42, 설계 및 건축 특징으로, 무균 제조의 완전성을 유지하기 위해 적절한 시설 설계 및 건축 특성의 중요성이 강조되었다.
두 번째로 자주 지적된 사항은 Section 211.113, 미생물 오염 관리에 대한 것으로, 생존 가능한 미생물 오염 물질을 적절히 관리할 필요성이 부각되었다.
마지막으로, 세 번째로 많이 지적된 사항은 Section 211.67, 장비 세척 및 유지보수와 관련된 것으로, 장비 세척 및 예방 유지보수가 부족한 점이 문제로 지적되었다. 이 기사에서는 FDA에서 발행한 Form 483을 바탕으로 2024년 감사 동향을 분석한다.
2024년에는 CFR Section 211.113, 211.42, 211.192, 211.100, 211.67, 211.160(b) 및 211.22(a)(d)가 무균 시설에 대한 전체 감사 결과의 거의 70%를 차지했다(그림 1). Section 211.113 및 211.42와 관련된 주요 지적 사항은 2023년 동향과 일치했다.
두 번째로 자주 지적된 CFR Section인 211.192는 생산 기록 검토 부족에 관한 것이다.
세 번째로 가장 빈번하게 관찰된 것은 Section 211.100, 서면 절차 및 일탈; 211.67, 장비 청소 및 유지 보수; 및 211.160(b), 일반 요구 사항, 실험실 관리. Section 211.22(a)(d)는 품질 관리 부서의 책임으로, 전체의 70%의 지적 사항을 차지한다.
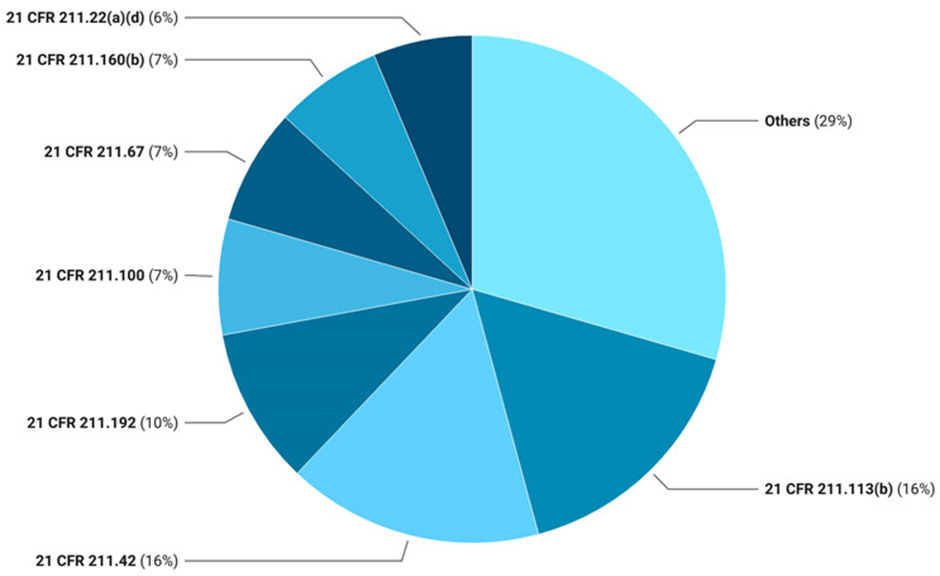
그림 1: 2024 파이 차트
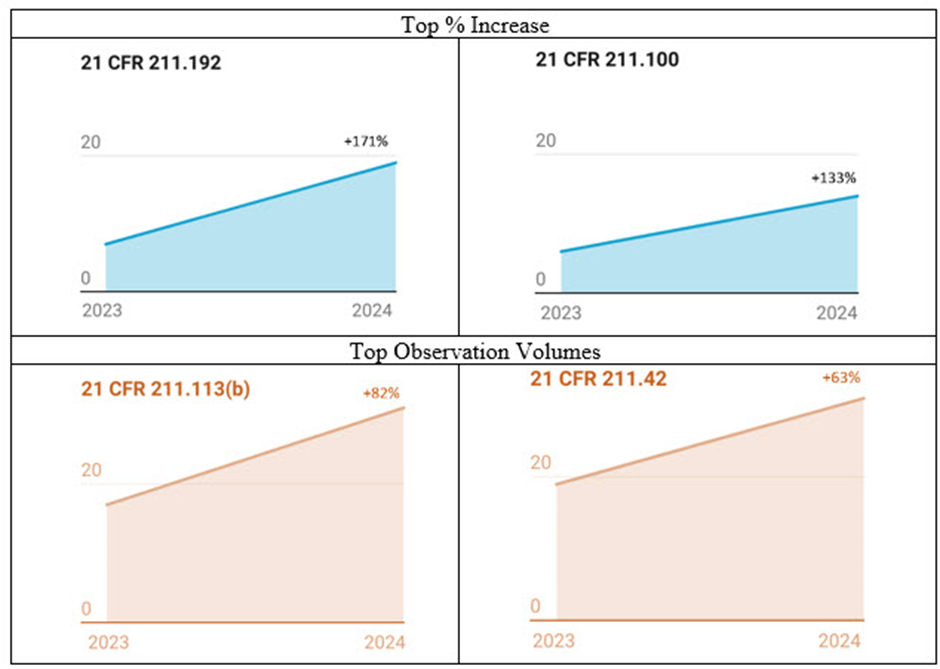
그림 2: 2023–24년 감사 지적 사항 증가율 및 건수. 숫자는 발표된 Form 483 감사 지적사항을 기준으로 한다.
2023년부터 2024년까지 지적 사항 증가율이 가장 높은 항목
CFR Section 211.192는 2024년 171% 증가하여 가장 높은 지적 사항 증가율을 보였다(그림 2).
이 Section에서는 교차 오염 조사를 포함한 조사 수행의 중요성을 강조한다. 배지 충전 실패 조사 관련 워닝레터에서 FDA는 회사가 청소한 작업자를 오염의 주원인으로 적절하게 고려하지 않았다고 지적했다. 또한, 규제 기관은 회사가 제조 중 유리 파손에 대한 적절한 개입을 하지 않았음을 확인했다.
또 다른 워닝레터 예시로, FDA는 이 회사가 완제품 테스트 중 두 번의 무균성 실패 사례를 겪었다고 강조했다. 오염된 미생물을 확인했지만, 근본 원인 조사에는 실패했다. 주요 주사제 제조업체에 전달된 지적 사항들은 조사 견고성이 저하했음을 나타낸다.
적절한 조사 도구 및 방법론에 접근할 수 있는 상태에서 기술적으로 자격을 갖춘 팀이 조사를 수행해야 한다.
첫 번째 단계는 무균 제조 조직이 고품질의 조사자 교육과 엄격한 적격성 평가 프로그램을 갖추고, 내부 역량을 검증하기 위한 독립적인 외부 전문가 인증 과정을 포함해야 한다.
2024년 두 번째로 높은 증가율을 보인 지적사항은 Section 211.100으로 133% 증가를 보였다.
해당 섹션의 대부분의 지적사항은 공정 밸리데이션과 관련되었으며, 특히 변동성(variability) 평가를 위한 지원 데이터 생성 부족이 주요 원인이었다.
이러한 지적사항의 증가는 FDA가 기존에 고형 제형(oral solid dosage) 제조소에 대한 변동성 관리뿐만 아니라, 무균 주사제(sterile injectables) 공정의 변동성 문제까지도 중점적으로 관리하고 있음을 시사한다.
지적사항에 대한 부적절한 대응이 워닝레터로 이어진 몇 가지 사례가 있었다.
한 사례에서 FDA는 워닝레터에서 제조 공정의 변동성(배치 내 및 배치 간 변동성)을 평가하는 지원 데이터가 부족하다고 지적했다. 위반 사항을 바탕으로 FDA는 전문 컨설턴트의 필요성을 강조했다.
이와 같은 규제 리스크를 최소화하려면, 기업은 PV Stage 1에서 CPP(Critical Process Parameters)가 체계적인 식별, 통계적으로 타당한 방법을 통한 CPP가 CQA(Critical Quality Attributes)에 미치는 영향 평가를 철저히 수행해야 한다. 또한, Stage 3에서 변동성을 감지하기 위한 모니터링 시스템을 운영함으로써, 규제 위반 위험을 크게 줄일 수 있다.
2024년에 가장 많은 인용 횟수를 기록한 지적사항
2024년 지적사항에서 FDA가 검사한 무균 주사 시설에 대한 지적사항 발생 건수는 2023년에 비해 483건으로 크게 증가했다. Section 211.113, 미생물 오염 관리 및 Section 211.42, 무균 시설 설계 및 건축 특징과 관련된 지적사항은 FDA로부터 지속적으로 많은 조사를 받았다.
Section 211.113과 관련된 워닝레터에서, FDA는 미생물 오염을 최소화하기 위한 작업을 모니터링하기로 했지만, FDA는 이를 충분한 조치로 인정하지 않았다. 해당 시설이 무균 작업자가 적절하게 무균 절차를 준수할 수 있도록 보장하는 방법이 명확히 제시되지 않았기 때문이었다.
FDA는 열악한 무균 작업 관행, 부적절한 가운 착용(gowning), 배지 충전 프로그램, 기류 시각화 연구 및 멸균 공정 적격성 평가를 지적했다. 이는 FDA가 강조해온 바와 같이, 환자 안전을 위한 자동화 및 아이솔레이터(isolator) 기술 도입을 장려한다.
무균 작업자의 행동 관찰(aseptic behavioral observation)의 증가는, RABS(Restricted Access Barrier System) 기술을 활용하여 다르게 생각해야 한다. 이를 위해, 산업에서 인정받은 교육 프로그램을 도입하고, 가상 현실(VR) 기술을 활용한 실용적이고 효과적인 교육 도구를 적용해야 한다. VR 기반 교육은 무균 작업자의 클린룸 청소, 설정 및 작업 과정에서의 주관적 판단 오류를 방지하고, 샘플링 및 무균 시험 수행 과정에서 공정의 완전성을 보장한다.
2024년, CFR 211.42, 무균 시설 설계 및 건축 특징과 관련된 지적사항도 상당히 많았다. 대다수는 설계 요소와 환경 모니터링(EM) 프로그램의 적절성과 관련이 있었다. Section 211.42 관련 워닝레터에서 FDA는 각 생산 배치와 관련하여 무균 처리 영역에서 비생물성 입자 모니터링, 작업자 모니터링, 활성 및 수동 생균 에어 샘플링 및 미생물에 대한 중요 표면 샘플링이 수행되지 않았다고 지적했다. 또한, 워닝레터에서 무균 제품이 제조되는 클린룸 세척 및 소독 공정을 검증하지 않았다는 점을 강조했다.
2024년에는 무균 작업 완전성 관련 지적사항이 있었다. 이러한 사례 중 하나로, FDA는 제조소가 비디오 검토를 포함하여 감독 하에 장비 세척, 소독 및 멸균이 수행될 것이라고 확인했음에도 불구하고, QA가 배치 출하 과정을 영상으로 검토하기에 비디오 보존 기간이 얼마나 충분한지에 대해 설명하지 않았기 때문에 대응이 부적절하다고 지적했다.
미비한 세척 밸리데이션 개선의 필요성도 나타났으며, 특히 독성 및 효능이 높으며, 세척 용제에서 낮은 용해도, 세척을 어렵게 만드는 특성, 적절한 스왑법 위치 및 최대 보존 시간 설정을 포함하는 모든 최악의 경우를 식별하고 평가하는 데 중점을 두었다. FDA는 이러한 지적사항을 해결하기 위해 해당 기업들의 주제별 지원이 필요하다고 판단했다.
2023년 및 2024년 제품 회수(리콜) 동향 및 분석
2023년 및 2024년 동안 제품 회수(리콜)에 대한 평가(그림 3)에 따르면, 미국에서 무균 의약품(바이알) 리콜의 상당 부분이 다음과 관련이 있는 것으로 나타났다.
• 이물질 혼입 - 주로 유리 입자 발견
• CQA 실패 - pH, 불순물 및 함량 시험(assay) 불량
• 무균 보증 실패 – 배지 충전 실패, 무균 시험 실패
• GMP 일탈 – 보관 및 운송 과정에서의 온도 일탈
• 라벨링(labeling) 규정 미준수 – 표시 오류
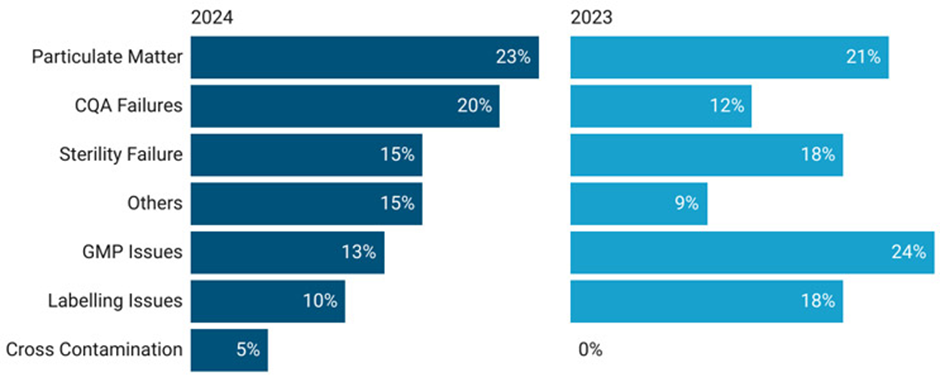
그림 3: 2023–24 제품 리콜(바이알) 동향
눈에 보이는 유리 입자 또는 유리 파손 관련 리콜 추세가 지속되고 있으며, 문제를 해결하기 위한 규제 기관의 노력에도 불구하고, 여전히 개선되고 있지 않다. 가능한 경우, 특히 최종 멸균 제품의 경우, 폴리프로필렌 바이알과 같은 대체 포장 기술의 원활하게 도입할 수 있도록 규제기관의 지속적인 지원이 필요하다. pre-gamma 멸균 형태로 폴리프로필렌 바이알이 제공될 수 있기 에 충전 및 완제 작업 중 오염을 초래할 수 있는 추가적인 취급 및 장시간 노출(바이알 세척 및 멸균 과정)을 생략할 수 있다. 멸균 백(sterile bags)에서 폴리프로필렌 바이알로의 전환은 환자 및 의료 종사자의 규정 준수와 안전성을 향상시킬 것이다.
또한 2024년에는 교차 오염과 관련된 리콜 사례도 보고되었다.
CCS는 Annex 1의 요구사항이지만 FDA 규제 시설에서도 중요하다. 제조 시설은 결함 요소와 환자에 미치는 영향을 결정하기 위해 업계 전문가의 지원을 받아 위험 평가를 수행하여 CCS를 재검토해야 한다. 이 선제적 조치와 필요 시 개선 과정이 향후 리콜 가능성을 최소화하고, 오염 관리 관련 감사 시 지적될 위험도 줄일 수 있다.
결론
2024년 FDA 483건의 지적사항 및 리콜 사례를 분석하면, 무균 시설에 대한 FDA의 기대 사항을 파악할 수 있으며 2025년에 규제 동향을 예측할 수 있다.
완전성 입증된 EM 프로그램, 무균 모니터링, 기존 공정 및 설계를 정당화하는 엄격한 오염 관리 위험 평가를 포함하는 완벽한 오염 관리 전략은 주로 환자 안전에 직접적인 영향을 미치기 때문에 모두 무균 작업에 필수적이다.
수기 배치 기록 문서화 방식으로는 더 이상 규제 기관의 요구를 충족할 수 없으며, FDA는 생균 및 비생균 오염으로부터 환자에게 미치는 영향이 큰 제품을 보호하기 위해 디지털 및 최신 제조 기술을 채택하도록 강력히 요구하고 있다. 규제 및 업계의 노력은 실제로 미국 시장에서 무균 의약품의 장기적인 공급 안정성(supplay security)이 확보될 것으로 기대된다.










